

丝状支原体( Mycoplasma mycoides ),地球上体积最小、结构最简单的生命形式之一。在这幅水彩画中,棕褐色的缠绕线条即是丝状支原体细菌染色体上的DNA;图片中间画有一个复制叉,DNA 聚合酶复合体(橙色物质)正在复制基因组,为最终的细胞分裂做准备;洋红色的结构是核糖体,信使 RNA 在这里被翻译成蛋白质;外层致密的深绿色绒毛代表连在细胞脂质膜外的碳水化合物链。下文中介绍的 WholeCell 计划模拟的生殖支原体(Mycoplasma genitalium),是比丝状支原体体积更小的一种支原体细菌。绘图:David S. Goodsell
(文 / Brian Hayes)差不多 30 年前,哈罗德•莫洛维兹(Harold J. Morowitz,美国生物物理学家)提出了一个对当时的分子生物学而言相当大胆的计划——研究地球上最小的单细胞生物体之一、一种支原体( Mycoplasma )细菌。他列出了这项行动计划的几个要点,第一步要做的,是解码这种细菌的完整基因序列,而这将反过来揭示出细胞中所有蛋白质的氨基酸序列。在上世纪 80 年代,读出整个基因组还不像如今这样稀松平常;但莫洛维兹论证说,只要基因组足够小,分析就是可能的。他计算出支原体 DNA 的信息量大约是 16 万字节,然后说:
换句话说,这么多的 DNA将编码出大约 600种蛋白质——可见生命的逻辑能在 600步以内写完。彻底理解一个原核细胞的运作机理是一个可见的概念,在可以实现的范畴之内。
莫洛维兹的计划中还有一点引人思考的地方:
在 600 步,写出一个计算机模型是可行的;而实验室里进行的每一项实验都可以在计算机上运行。直至这些对应抵得上分子生物学范式的完整性。
在如今这个基因组学和蛋白质组学都已如产业规模的现代,回过头去看这些建议,无疑莫洛维兹在收集基因序列数据的可行性这一点是说对了。另一方面,在 600 步之内写下 “生命的逻辑” 和 “完全理解” 一个活细胞这两个挑战,看起来仍然相当艰巨。那么,制作出一个足以媲美真正生物体运作机理的计算机程序又如何呢?
碰巧的是,2011 年夏天,斯坦福大学的马库斯•克维尔特(Markus W. Covert)等9人,发表了一个名叫 “WholeCell”(全细胞)的计算机程序,实现的正是模拟 莫洛维兹当时提议的一种支原体属细菌——生殖支原体( Mycoplasma genitalium )的整个生命周期。模型中包含了所有主要的生命过程:DNA 转录成 RNA、RNA 翻译成蛋白质、营养素代谢产生能量和结构成分、基因组的复制,以及最终通过细胞分裂进行繁殖。
计算机模拟的结果看来确实与实验结果相一致。所以,现在不得不去面对这样一个问题:人类即将 “补完” 分子生物学了吗?
支原体,最小的生命形式
科学家做这类实验爱用支原体,是因为它们是最小的、并且可以说是最简单的具有自我繁殖能力的细胞。 (病毒的体积更小,但只能通过劫持宿主细胞的生化机制来进行繁殖。)
19 世纪时,人们第一次观察到支原体,当时以为看见了一种真菌。现在,支原体在分类学上被划归于微生物门下,但却是细菌王国中的异类。它们没有包裹在其他微生物之外的刚性细胞壁,只有一个脂质膜。其结果之一便是支原体对多种抗生素都具有耐药性,尤其是那些通过干扰细胞壁合成而起效的药物。支原体是多种人类疾病和其他动物疾病和植物疾病的病因。或许最有名的一种人类疾病,是有时候被称为 “非典型肺炎” 的肺部感染。
克维尔特小组计算机模拟的生命——生殖支原体,自 1980 年以来才为科学界所知,当时是从几名尿道炎患者身上分离而来。即便在支原体当中,生殖支原体的个头儿也是小的,细胞直径大约为 0.5 微米。相较之下,另一种更为知名的细菌大肠杆菌( Escherichia coli ),长有 2 微米,体积差不多是前者的 50 倍。生殖支原体在遗传信息的量上来说也很小:它唯一的环状双链 DNA 上只有 580076 个碱基对和区区 525 个已识别的基因(这比莫洛维兹估计的还要少)。相比之下,大肠杆菌的基因组约有 460 万个碱基对,编码 4300 个基因。
紧凑的细胞和精简的基因组使支原体不仅是测试软件时有用实验台,在探索维持生命所需的最小设备——“湿件”(wetware)而言也一样。2010年,克雷格•文特尔研究所(J. Craig Venter Institute)的克雷格•文特尔(J. Craig Venter)、克莱德•赫钦森(Clyde A. Hutchison III)和汉密尔顿•史密斯(Hamilton O. Smith)等人发表了一个著名的实验。他们对一个丝状支原体的基因组( Mycoplasma mycoides )进行了测序,将测得的碱基序列储存为计算机文件;然后,做了一些编辑以形成一个可识别的 “水印”,再根据改变后的序列合成了 DNA。最后,困难的地方来了,他们把人造 DNA 嵌入另一种支原体的细胞里,取代了原生的遗传物质。改造后的细胞继续生长,并完全在人工基因组的指引下进行繁殖。这个实验可以被视为人类向创造完整的合成生命形式迈出的一步。
在某些方面,用电子计算机模拟生命比通过化学成分合成要难。材料对了,生物学家在没有完全了解各部分相互作用细节的情况下,是有可能组装出一个活细胞来的。但是,计算机程序员必须描述清楚每一个分子事件才行。
建立计算机模型,模拟生命
在建立计算机模型时,需要做出大量的选择和妥协以找到适当的具体程度。以碳水化合物的代谢为例,其中糖(如葡萄糖)分解产生水和二氧化碳。在最抽象的层面上,这个过程变成一个化学方程式:
C6H12O6 + 6O2 →6CO2 + 6H2O
而这个式子并没怎么反映出细胞里实际发生的事情。进一步分析会多出几十个中间步骤。例如,六碳的葡萄糖分子首先被分成两个三碳的丙酮酸分子,释放出的能量被捕获后以高能磷酸键的形式贮存在三磷酸腺苷(ATP)里。
这样不断地细分下去,最终就会形成一张巨大的化学反应网,像已故的唐纳德•尼科尔森(Donald E. Nicholson)设计的那张著名的代谢途径图。而且,到了这一步之后还可以细分。在原则上,模拟可以跟随每一个分子——或者从这个意义上说,每一个原子——在细胞机器中穿行的轨迹。金发姑娘策略(The Goldilocks strategy),便是在平淡无奇的抽象与毫无意义的逼真之间寻求一条中间的道路。
WholeCell 计划的作者选择对模型的不同部分采用不同的具体程度。他们把某些关键的大分子表示为独特的、可识别的实体。较小的分子则被视为聚合量,程序追踪其数目,但不对个体进行标记。
这两种表现模式之间的区别,可以在模型中负责蛋白质合成的那部分清楚地看到。核糖体是细胞中合成蛋白质的场所,这种大的细胞器被记为个体;每一个核糖体都有自己的身份和历史,计算机程序给每一个核糖体都配置了单独的内存。但是,构成蛋白质的元件——氨基酸分子,程序并没有单独标记。模型只会追踪每种类型的氨基酸的总量:有一个变量计算所有的丙氨酸分子,另一个计算所有的赖氨酸分子,以此类推。
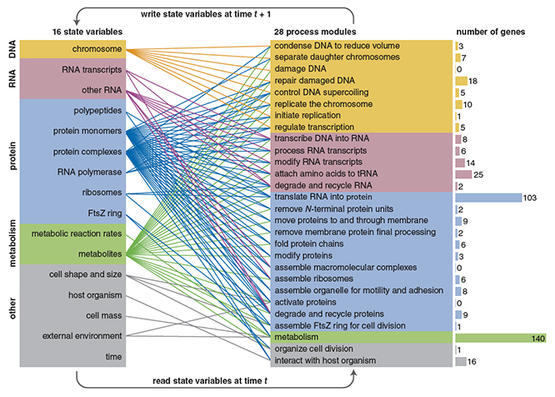
WholeCell 模型一共有 28 个处理模块,分别对应于主要的细胞活动,如基因组的复制、蛋白质的合成以及受损 DNA 的修复。此外,还有 16 个状态变量,负责记录各个子系统在每一个瞬间所处的状态。启动时,程序先将各个状态变量的值进行初始化,使其与细胞分裂后产生的 “新生” 细胞相当。接下来,所有的 28 个处理模块运行一秒钟的模拟时间。在这段时间间隔之后,状态变量的值会被计算结果更新,然后重复这一循环。模拟程序继续运行,直到细胞完成生长和分裂。对生殖支原体而言,一代的时间通常是 9 个小时,也即模拟循环重复大约 3.2 万次。程序的运行时间与生殖支原体过完一代的时间基本相等。制图:Brain Hayes
在 WholeCell 计划的网页上可以找到 这一模拟程序的源代码,用 MATLAB 语言写成。还有一个知识库,里面有用于构建该模型的定量信息。
加上克维尔特,该软件的主要作者还包括克维尔特小组的两名研究生卡尔(Jonathan R. Karr)和桑维(Jayodita C. Sanghv)。描述模型的研究报告2011年7月在《细胞》杂志发表,报告的作者除了卡尔、桑维和克维尔特之外,还有斯坦福大学的 Derek N. Macklin、Miriam V. Gutschow、 Jared M. Jacobs 和 Benjamin Bolival Jr.,以及文特尔研究所的 Nacyra Assad-Garcia 和 John I. Glass。
WholeCell 模拟程序:模块与策略分析
为了理解全细胞模拟程序是如何工作的,我选择了三个模块仔细考察,分别是新陈代谢模块、遗传信息转录模块和分管细胞生长的大小和形状的模块。
新陈代谢模块:动力室
新陈代谢模块是大部分经典的生化代谢反应发生的地方。这里处于细胞经济体的蓝领阶层,与能源生产、加工制造、原材料和废物处理打交道。(其他模块则更多是关注信息处理的白领事务。)
即使在一个微小如生殖支原体的细胞里,代谢过程也牵涉到庞杂而错综的化学反应。WholeCell 模拟模型的代谢模块包含了104 种酶、585 种基板、441 个化学反应和 204 次运输过程。
代谢网络的大小和复杂程度使很多常规的分析方法都派不上用场。任意一个化学反应的速率都部分取决于反应物和生成物的浓度。但是,在这里一个反应的生成物同时又是另一个反应的反应物,所有的过程都紧密结合,不能单独解决。更为复杂的是,生物网络中含有物质的循环,比如碳水化合物代谢的柠檬酸的循环。有了循环,任意一个反应的生成物都可以绕全程一圈,然后作为同一个反应的反应物再次加入这个循环,从而使流经网络的物质总量无法被唯一地定义。
为了回避这些困难,WholeCell 的代谢模块采取了一个叫通量平衡分析(flux-balance analysis)的方法。其基本思想是,就算一个反应网络没有 唯一 的解,但 最佳 的解还是可以有的。找出这个最佳解的过程,很像是在优化化工厂或炼油厂日常运营时用到的算法。假设一家炼油厂有一系列产品(汽油、柴油等等),其制造成本和市场售价各不相同。用线性规划的数学方法能计算出利润最大时的产品结构。将同样的方法应用到活细胞的代谢中,也能得到一组使可用资源(如营养物)的有效利用率最大的反应速率。(目前还不能肯定微生物是否以这种方式优化其生长,但在达尔文的自然选择理论下,这是一种合理的假设。)
转录模块:遗传信息复制
转录模块中的计算与代谢模块有很大的不同。之前的是用线性规划,现在则是概率控制下的离散事件系统。
遗传信息的转录,从 RNA 聚合酶的分子与染色体上一处叫启动子的位点相结合开始。接下来RNA 聚合酶会绕着双螺旋结构制造出一个单链信使 RNA,其碱基序列与 DNA 链中的一条完全互补。转录结束后,聚合酶会从 DNA 链上脱落,放开 RNA。转录过程中每一步都需要很多其他分子的参与,比如起始因子、延伸因子、终止释放因子和提供能量的物质,还需要额外的核苷酸补充到增长的 RNA 链中。
WholeCell 系统中,每个 RNA 聚合酶分子都是一个单独的对象,有四种可能的状态:①转录,②与 DNA 的启动子相结合,③与 DNA 的其他部分结合,④游离状态。这四种状态之间的转换是随机事件,其发生概率经计算与实验观察到的分布结果一致。另外,不同的启动子位点与 RNA 聚合酶的亲和力不同,所以有些位置 RNA 聚合酶分子处于结合状态的概率比其他地方要高。
由于有了这些概率事件,WholeCell 模型中就存在不确定性因素。可以预想得到,即使初始条件和环境都相同,每一次运行都会产生稍微不同的结果。但话说回来,波动和偶然事件在现实的生物学中也会发生;就算是完美的克隆体也不会完全遵循同样的轨迹走完各自的一生。
在阅读转录模块源代码的过程中,我注意到了一些值得注意的地方。假设掷一次数字骰子的结果,决定了一个特定的 RNA 聚合酶分子将与一个启动子位点相结合;那么,要是这时所有的启动子位点都已经被占了会如何?如果两个聚合酶分子试图在同一时间与同一个启动子位相结合,会发生什么事情?如果两个转录酶在各自沿 DNA 链移动的过程中撞到一起了又该怎么办?大自然似乎能够自然而然地解决这些矛盾,但建模者则必须把所有的一切都想清楚才行。
酶在沿着染色体凿沉的过程中发生碰撞并不是罕见的事件。WholeCell 的模拟结果表明这类碰撞每秒钟发生一次,在一个完整的细胞周期中或许会发生 3 万次。因此,模型需要制定规则来决定谁走谁留。
生长模块:细胞的大小和形状
WholeCell 模型并没有在空间组织的有关细节上面下很大功夫。代谢模块把细胞看做一个充分搅拌后的容器,里面所有的分子不论位置如何,相互反应的机会都是相同的。转录酶和复制酶虽然与细菌染色体的特定位点相结合,但其坐标却是一维的,根据线性的基因序列来定位;它们并没有确定在三维空间中的位置。
不过,在这个模拟程序里还是包含了一个表示细胞几何形状的状态变量,这个变量描述了细菌的形状和最终的分裂。让人感到奇怪的是,模拟模型定义的形状并不是细胞实际的样子。生殖支原体通常被描述为梨形或者烧瓶一样的形状——一个突出来一截的球。将这个细节纳入模型不具任何生物学的意义,只会徒增复杂程度,因此模拟细胞被设定成更简单的几何形状。它最开始是一个小球,而后伸长成圆柱体,上下底面呈半球状。细胞周期结束后,两条复制的染色体迁移到细胞的两端,圆柱的中间开始收缩、断裂,最后形成两个新的细胞。
细胞生长的原理并不难懂:随着细胞质的体积不断增加,包裹整个细胞质的细胞膜的表面积也必须增大相应的量。细胞分裂的机制则更为复杂,还有无法解释的地方;但 WholeCell 模型还是给出了一个初步的描述。其中关键的成分是一种叫 FtsZ 的蛋白质,它会形成一个环,箍住生长中的细胞,不断收紧,最终把细胞一分为二。
WholeCell 模型所基于的数据,是从 900 种出版物中收集来的。从这些资料中提取出的大约 1900 个数值最终成了模型的参数。这是一个了不起的工作,让模拟立足于真实的数据。
然而,参数多达 1900 个本身也是个问题。假设每个参数都相当于一个控制旋钮,转动它就能调节模型的行为,那么摆弄足够多的旋钮,就能使输出“匹配”几乎任何想要的结果。当我向克维尔特提起这个问题时,他随即引用约翰•冯•诺伊曼的一句话,“给我 4 个参数,我能画出一头大象,给我 5 个我能让它翘起鼻子来”。不过克维尔特接着又说,WholeCell 模型中的 1900 个参数既没有被摆弄也没被拿去翘鼻,几乎所有的值都直接取自实验测得的结果。它们制约了模型,而不是把模型调整到一个设定好的输出上面。
可有了这些还不够。模型使用的数据来自许多不同的实验,这些实验由不同的人员进行,时间跨度有几十年。有很多参数的值都取自生殖支原体以外的生物,纯粹是因为关于支原体的生理知识有很多都还没弄清楚。有了这些零散的数据来源,各个参数的测量结果并不总是一致也就毫不奇怪了。例如,一份细胞质的成分表中(由莫洛维兹 50 年前发表)说支原体只含有微量的半胱氨酸,而基因组的分析结果表明支原体蛋白所需要的半胱氨酸含量要高很多。要成功模拟细胞,就必须协调这些不一致。
克维尔特和他的同事通过制定系统的限制,然后寻找满足约束条件的参数值,同时尽可能少地偏离测量值,最终解决了这个问题。起初他们试过用正规的优化算法,但都没有得出一个可行的解决方案。于是,他们采用了一种启发式求解法,从那些看上去最可靠的参数算起。在得出更完整和更准确的生化数据之前,一些这样的妥协仍将是必要的。
与此同时,发表在《细胞》上的那篇论文确实给出了在生理学上站得住脚的模拟结果。细胞周期的持续时间、生物量的生长率和各种代谢产物的浓度,所有的结果都相当接近于真实测定的数值。一系列的 “基因剔除” 实验(从染色体上去除单个基因然后看运行结果,如果失去一个基因后模型无法运行,则说明这个基因是必须的),进一步支持了该模型的鲁棒性。在模拟结果与活体实验数据相一致的情况下,79% 的基因都是必不可少的。
还有一个发现,让我们对一种已知的现象有了更好的理解。支原体的细胞周期中,基因组复制的早期阶段是由转录酶和启动子结合的启动阶段,接下来才是基因组的复制。这些早期阶段和后期阶段耗时有长有短,但其总和、也即整个细胞周期的时间,则相对极少变化。仔细考查模拟模型的内部细节后,我发现了这一奇怪现象的原因。合成新的染色体需要用到核苷酸,而核苷酸的制造贯穿于整个细胞周期。如果基因组复制的早期阶段很短,那么后期复制的速率就会由于缺乏核苷酸而减慢;而如果早期用时较长,后期就有充足的核苷酸支持全速复制。
制造人造生命形式,无论是用软件还是在合成的细胞质之中,这个想法一直以来都备受争议。将近 200 年前,玛丽•雪莱(Mary Shelley)写下了一篇深刻思索这一主题的文章——《弗兰肯斯坦》(Frankenstein, or the Modern Prometheus)。在雪莱的时代,有关人造生命的辩论还囿于活力论与机械论之争。活力论的拥护者认为,生物与无机物的区别在于生物体内有某种 “生命花火” 或者赋予活力的原理。而反方的机械论则得到了笛卡尔的支持——他将动物比作上了自动发条的机器。
在科学的范畴内,活力论的学说早就死了;但对于生命可以通过拆解有机体并将其各组成部分编目这种方式被彻底了解,人们仍旧心存抵触。在分子生物学刚刚起步,还急于突进的时期,DNA 被视为 “生命的蓝图”,是建造活细胞的全套指南。生命的核心过程被看成符号的操控——怎么把 G 和 C、A 和 T 配对,然后把 4 个字母写就的核苷酸映射到氨基酸的 20 种类别里。只要能看懂这份蓝图并破译遗传信息,人类就能了解生命运作的所有道理。而如今,我们已经能非常流利地读出 DNA 序列,有一个事实也变得越来越清晰——生命中含有比分子生物学的 “中心法则” 更多的东西。
用计算机程序模拟一个活细胞的想法,正好处在了一场争论的交火地带,一边是简化论,而另一边则是一个更加综合的生物学视野。一方面,WholeCell 计划无比清晰地表明,DNA 序列本身并非解开生命之锁的关键。虽然从 DNA 到 RNA 到蛋白质的信息传输是模型的核心要素,但这一过程并不是只是一一对应那么简单。重点是分子,而不是符号。
另一方面,尝试建立这样一个模型,其行为本身便宣告了生命可以被理解,不存在所谓的超自然元素,生命可以简化成一个算法、一个有限的计算过程。模拟细胞里发生的一切都遵循可以被人枚举和理解的规则,原因很简单,这些规则是我们写就的。
我非常期望模拟生物学在技术的成功会形成一种新的合成,封住围绕这些问题的哲学争论。对此我不抱太大的期望。
参考文献
- Barile, M. F., and S. Razin (eds). 1989. The Mycoplasmas . New York: Academic Press.
- Bonarius, H. P. J., G. Schmid and J. Tramper. 1997. Flux analysis of underdetermined metabolic networks: The quest for the missing constraints. Trends in Biotechnology 15:308–314.
- Brenner, S. 2010. Sequences and consequences. Philosophical Transactions of the Royal Society of London 365:207–212.
- Covert, M. W., et al. 2001. Metabolic modeling of microbial strains in silico. Trends in Biochemical Sciences 26:179–186.
- Covert, M. W., N. Xiao, T. J. Chen and J. R. Karr. 2008. Integrating metabolic, transcriptional regulatory and signal transduction models in Escherichia coli. Bioinformatics 24:2044–2050.
- Crick, F. H. C. 1973. Project K: The complete solution of E. coli. Perspectives in Biology and Medicine 17:67–70.
- Gibson, D. G., et al. 2010. Creation of a bacterial cell controlled by a chemically synthesized genome. Science 329:52–56.
- Karr, J. R., et al. 2012. A whole-cell computational model predicts phenotype from genotype. Cell 150:389–401.
- Lerman, J. A., et al. 2012. In silico method for modelling metabolism and gene product expression at genome scale. Nature Communications 3:929.
- Loeb, J. 1912. The Mechanistic Conception of Life: Biological Essays . Chicago: University of Chicago Press.
- Morowitz, H. J. 1984. The completeness of molecular biology. Israel Journal of Medical Sciences 20:750–753.
- Orth, J. D., I. Thiele and B. Ø. Palsson. 2010. What is flux balance analysis? Nature Biotechnology 28:245–248.
- Suthers, P. F., et al. 2009. A genome-scale metabolic reconstruction of Mycoplasma genitalium , iPS189. PLoS Computational Biology 5:e1000285.
- Tomita, M., et al. 1999. E-CELL: Software environment for whole-cell simulation. Bioinformatics 15:72–84.
编译自: 《美国科学家》,Imitation of Life
文章图片:americanscientist.org;(小图)v.youku.com