
我们都知道健康人具有两套染色体组,染色体的两个拷贝其中一条来自父亲,另一条来自母亲。先前的技术已经可以对人类的整套基因组进行测定,却无法对这两个拷贝加以分辨。现在,在11月3日发表的《自然-生物技术》上,来自加州大学圣地亚哥医学院路德维希癌症研究所的一个团队解决了这一问题。
人类繁衍至今,我们祖先的染色体片段早已在无数代人的繁殖过程中因基因重组而打乱。那些整体遗传自父本或母本、未被重组打断的基因区段,被称为单体型。在任兵教授的领导下,路德维希癌症研究所开发出一种被称为“HaploSeq”的新技术。通过结合分子生物学与生物信息学的方法,HaploSeq技术使得研究人员得以快速确定发生在同一段染色体上的基因变异,从而确定单体型(Haplotype)。
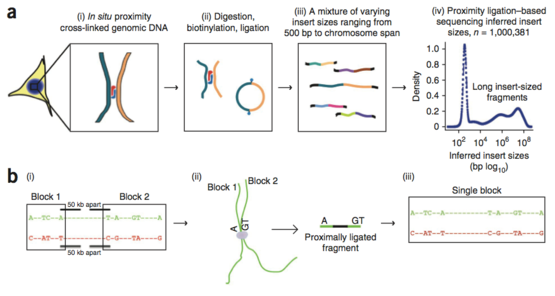 HaploSeq工作原理示意图:(a)邻近式连接实验:细胞内交联在一起的DNA链被片段化后连接,并从细胞中提取出来进行测序。(b)利用测序结果构建单体型:用短序列构建单体型域,并将各个单体型域串联,得到跨染色体的单体型。图片来源: Siddarth Selvaraj et al.Nature Biotechnology. 2013.
HaploSeq工作原理示意图:(a)邻近式连接实验:细胞内交联在一起的DNA链被片段化后连接,并从细胞中提取出来进行测序。(b)利用测序结果构建单体型:用短序列构建单体型域,并将各个单体型域串联,得到跨染色体的单体型。图片来源: Siddarth Selvaraj et al.Nature Biotechnology. 2013.
研究者首先用邻近式连接测序技术对基因组进行测序。交联的DNA链首先被限制酶切断,再被连成人工片段。这些人工片段包含了一些系列不同长度的插入片段,大小从500个碱基对到数千万碱基对不等。对短的片段进行分析能够得出许多小的单体型域,而长片段则能将这些单体型域串联起来,区分出单体型。当测序覆盖率达到一定程度,研究者就能够将所有不连贯的单体型域整合成一个单体型——也就是遗传自母亲或者父亲的基因型。任兵表示:“该技术使得临床医生能够更好的评估个体的疾病风险,以后可能会为个性化医疗带来变革。”
任兵指出,由于HaploSeq建立在传统的测序方法上,所以他预计这一技术更容易被临床人员广泛应用。这一技术最直接的应用,便是帮助临床医生评估一个人罹患某种特定疾病的风险。举例来说,癌症易患人群通常带有多个DNA突变,而HaploSeq技术可以确定这些突变缺陷是否位于同一条染色体。如果是,那么另一条正常的染色体则会补偿这些突变使得个体患癌症的风险降低。反之,这些突变可能导致癌症。
此外,进一步的研究将使利用该技术改善当前器官匹配的流程成为可能。研究人员希望这些技术能够通过评估器官捐赠供体与受体基因组的匹配程度,确定两套基因组的兼容性。“进行这种应用前仍需要更多的研究。我们需要创建一个基因组数据库,来更好的帮助受助者与捐助者。”任兵说。
再者,在遗传研究领域,该技术将帮助研究者们分析人类的迁徙状况,并根据基因组的差异来判断他们的祖先。任兵教授表示:“HaploSeq可以通过比较你和你邻居的DNA序列来判断你们是否有共同的祖先。类似这样,利用这一技术,我们可以研究每一个单独个体及他们之间的联系。而随着数据样本的增大,个体之间的联系将被更为精确的确定。”这样的研究将对HapMap计划提供支持——这是一个目前正在进行的国际项目,旨在评估全球人类遗传变异的情况。
信息来源:EurekAlert!
文章题图:weibo.com
相关的果壳网小组