
5月25日,《自然-生物技术》发表了来自华大基因的炎黄一号单倍型组装结果 。
2008年,炎黄一号一诞生即成为“亚洲第一”——第一个蒙古人种的全基因组测序结果。经过科学家的不懈打磨,到《自然-生物技术》这篇最新论文发表的此刻,从测序数据质量上来说,炎黄一号终于成了“世界第一”!
作为和这项研究有些渊源的基因组学工作者,笔者将尽我所能,解读炎黄一号基因组的系列科研和技术进展。以期让大家理解,个人全基因组测序现在究竟进展到了什么地步?这项工作的意义和价值又在哪里?
基因组数据:能用,但还不够好
在这个“千元基因组时代”,个人基因组测序似乎已成了大众消费品。那么,为什么世界顶级的基因组研究机构和测序仪生产商,还在不断的测序更多的个人基因组,还要将个人基因组研究做到目前技术条件下“极致精美”的程度呢?
简单的答案就是,因为目前的基因组数据还不够好,因而也不够好用。
我们研究人类基因组最关心的问题,就是将染色体上所有的基因序列都测序出来,并且定位清楚,简单的说,目标就是将人类基因组从第一个碱基开始,一直到最后一个碱基结束,真正完整的呈现出来。
在基因组研究领域,人们对数据的可信度有一个基本的要求:单个碱基越准确越好,对单个碱基的覆盖深度越多倍越好,对整个基因组测得越完整越好,测序的“缺口(Gap)”越少越好。
以这些标准看,目前的基因组测序结果,还没有一个是完美的。
人类基因组计划:曾经的“最好”
自从人类基因组计划和科学狂人克雷格· 文特尔(Craig Venter)先后公布人类基因组图谱以来,基因组研究进入了全新的纪元。然而,这份图谱只是张“不够完美的参考图”,科学家们很快认识到,我们需要更多人的基因组, 才能真正将遗传与基因组信息应用到健康和临床领域。
但是因为测序基因组太过昂贵,科学家们选择了折衷的思路,那就是后来启动的国际人类基因组单体型图计划( HapMap Project),旨在了解人类遗传的单倍型和单点突变。虽然取得了一定的进展,但是根本问题仍然存在——测序的人类基因组数据太少,质量还不够好。
再后来,454公司测序了诺奖得主詹姆斯·沃森(James Watson)的基因组,并将其公布,但是沃森的基因组从测序质量上来讲,跟人类基因组计划公布的还不在一个水平上,所以,大部分科研工作者,还是在使用人类基因组计划所公布的基因组数据作为参考序列。
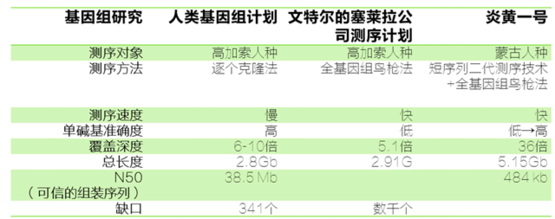
在2004年公布的人类基因组计划的数据中,对单个碱基的覆盖深度是6~10倍的覆盖深度,当时计算的人类基因组总长度约为2.8G,有341个缺口, N50(可信的组装测序序列)的长度为38.5 Mb,这个长度是人类基因典型长度的1,000倍,应该说,在当时的测序条件下,这样的数据已经是非常好的结果了。早两年文特尔公布的基因组覆盖度为5.1倍,基因组的总长度是2.91G,从2001年发表的那个版本看,缺口的数量有数千个,所以从测序数据质量上来讲,较人类基因组计划还是有一定差距,且当时人类基因组计划用的是“逐个克隆法(Clone by Clone)”的定位方法测序,这种方法前期需要大量的工作进行克隆的定位,因此很费时间,而后的测序和分析则相对容易。而文特尔采用“全基因组鸟枪法”测序,这种方法不需要大量的克隆定位,但对用来组装的计算机硬件软件要求很高,且容易出错,好处在于节省了时间,提高了效率。

文特尔(左)和负责“人类基因组计划“的科林斯(右),并肩开启基因组时代的双雄人物 图片来源:time.com
单就数据质量来说,人类基因组计划所得到的基因组图谱还是更加准确可靠。
但从上面的描述,我们可以看出几个问题,因为采取的基因组测序策略不同,公布的两个基因组,长度不一,缺口的数量不一,测序的质量也不一,从精益求精的角度看,二者都不够完美。
人类基因组:缺点在哪里?
首先,人类基因组还不够精确。人是“二倍体”,也就是有一半遗传物质来自父亲,一半遗传物质来自母亲,且在受精卵形成过程中,还会发生基因重组,这是人类遗传多样性的来源之一。科学家们需要更精确的“单倍型”数据,这样基因组才够“完美”,而这种“完美”正是研究者们追求的目标。
其次,人类基因组还不够多元。
按照传统的人种分类,人类按照肤色黑白黄棕,被粗分为四大类:尼格罗人种、高加索人种、蒙古人种、澳大利亚人种。基因组测序数据是从高加索人种开始的,人类基因组计划是人类的标准参考基因组,也是高加索人种的标准参考基因组。文特尔的基因组,测序对象是他自己,同样是高加索人种。
然而,从基因组研究的角度,为了尽可能地包括各种遗传背景,需要为更多族裔建立自己的参考基因组。
第一个蒙古人种基因组,正是由华大基因团队测序完成。2008年他们在《自然》杂志发表了《一个亚洲人的二倍体基因组测序(The diploid genome sequence of an Asian individual)》, 这就是我们俗称的炎黄一号。同时发表的还包括来自尼格罗人种的全基因组测序数据。至此,三种肤色人种的基因组数据总算凑齐了。
2008年的华大论文中,蒙古人种基因组的覆盖深度是36倍,看似比人类基因组计划的10倍覆盖度要高出很多,事实上,蒙古人种基因组测序采用的是短序列二代测序技术,而人类基因组计划和文特尔的基因组采用的是一代测序的长序列测序。虽然炎黄一号也进行了组装,但是非常难以达到人类基因组计划的水平,而且当时二代测序技术准确度难以跟一代测序技术相媲美,所以测序质量也比较低。需要更高的覆盖深度来弥补。
这些技术细节可以简单理解成,虽然蒙古人种基因组的覆盖深度高,但是由于序列的定位可能出现问题,且质量不够高,所以,达到的效果跟10倍覆盖深度的人类基因组计划那个标准图谱在单碱基的准确度上相差不大,从结构变异和单倍型角度看,可能还有一些差距。当然,从技术角度,两套基因组测序都“不够完美”,所以不好下定论,得具体数据具体比较。
炎黄一号:九年磨砺,成就“最好”
自炎黄一号的第一阶段结果发表以来,华大基因的科技工作者就开始不断完善“蒙古人种”的基因组图谱。
首先是2009年,华大基因与合作单位的一群年轻研究者在《自然-生物技术》发表了研究论文《构建人类泛基因组序列图谱(Building the sequence map of the human pan-genome)》。利用组装的方法构建出炎黄一号独有的大约5M的基因序列,并且验证了其存在并预测了其功能,而且将炎黄一号的基因组组装提升到新的水平。
2011年,华大在《自然-生物技术》上又发表了一篇新论文《Structural variation in two human genomes mapped at single-nucleotide resolution by whole genome de novo assembly》,将炎黄一号的新组装结果与另外一个尼格罗人种的组装结果进行比对,在原有基础上,发现了277,243个新的基因组“结构变异”,同时还发布了为此开发的新的组装流程。
我们可以这么理解这项研究,2008年版本无法发现的基因组结构变异,可以通过2011年的新方法找到,特别是小范围的(≤50 碱基对)和中等范围的(51~200 碱基对)结构变异。因为炎黄一号测的是短序列,所以确实比较难发现大的结构变异(>200 碱基对),这一问题一直困扰着通过短序列高通量测序进行基因组研究的研究者。
2015年5月,华大基因在《自然-生物技术》上发表了《De novo assembly of a haplotype-resolved human genome》,通过全基因组鸟枪测序法(WGS)结合全新策略(Fosmid-pooling)的分级组装方法,以及之前的二代测序短序列组装出人类单倍体水平的二倍体基因组。组装出了5.15Gb的二倍体基因组,单倍型的可信N50 为484 kb,还发现了7.49 Mb的独有序列。至此,历时9年的炎黄一号基因组已经趋于“完美”,这已是领先于目前已知的所有的人类基因组测序结果的最为完整的基因组图谱。
从2006年炎黄一号项目正式启动,到2015年5月25日《自然-生物技术》的最新论文发表。可以说,此时此刻,亚洲人在这一领域,真正领先于世界!
完美的基因组参考序列,意义何在?
早在十年前,詹姆斯·沃森就敢于发出宣告,“未来所有生物学只有以基因组开始才有希望发展! ”
当初的预言,正在一步步变为现实。
一个真正完美的基因组,意义相当深远。
首先,它对遗传学研究至关重要。通过炎黄一号组装的单倍型图,我们可以更为清晰地了解不同基因型之间的连锁关系、遗传特征,进而深入研究基因组重组的机制,基因组的各种修饰与不同单倍型的关系,基因组单倍型结构与基因表达、调控、修饰的关系等等。
热门的表观遗传学也离不开基因组信息。2010年,华大的研究者在《Plos Biology》上发表过炎黄一号志愿者的外周血单核细胞DNA甲基化图谱,该图谱的分析是基于之前组装的版本,相信在新的组装版本的推动下,类似研究将更加深入准确。随着下一步研究延伸至志愿者的各种组织(如肌肉、皮肤等),我们将对DNA甲基化的机制和组织特异性的甲基化基因表达有更加深刻的理解。
而对于目前火热的“精准医疗”和“个体化医学”而言,准确的基因组参考序列,将帮助真正“精准”的基因组数据分析。值得庆幸的是,有炎黄一号作为蒙古人种的参考序列,我们将离“精准”的目标更加接近。(编辑:游识猷)
参考文献
1. International Human Genome Sequencing, C. (2004). "Finishing the euchromatic sequence of the human genome." Nature 431(7011): 931-945.
2. Venter, J. C., et al. (2001). "The sequence of the human genome." Science 291(5507): 1304-1351.
3. Wang, J., et al. (2008). "The diploid genome sequence of an Asian individual." Nature 456(7218): 60-65.
4. Li, R., et al. (2010). "Building the sequence map of the human pan-genome." Nat Biotechnol 28(1): 57-63.
5. Li, Y., et al. (2011). "Structural variation in two human genomes mapped at single-nucleotide resolution by whole genome de novo assembly." Nat Biotechnol 29(8): 723-730.
6. Cao, H., et al. (2015). "De novo assembly of a haplotype-resolved human genome." Nat Biotechnol.
7. Li, Y., et al. (2010). "The DNA methylome of human peripheral blood mononuclear cells." PLoS Biol 8(11): e1000533.